")
The EU Clinical Trial Regulation (EU CTR 536/2014) is a comprehensive framework designed to enhance the safety, transparency, and efficiency of clinical trials conducted across the European Union.
A key aspect of this regulation is its emphasis on clear, accurate communication to ensure that all stakeholders, including trial participants and regulatory authorities, fully understand trial-related information.
To ensure this, the EU CTR sets strict translation requirements that sponsors and trial operators must comply with in every member state involved.
This blog highlights the reasons behind the transition from EU CTD to EU CTR, key changes, translation requirements, and the best practices for clinical trial translations.
What is EU CTR?
The EU Clinical Trials Regulation (EU CTR) (Regulation (EU) No. 536/2014) is the legal framework governing the conduct of clinical trials of investigational medicinal products within the European Union.
It replaced the previous Clinical Trials Directive (2001/20/EC) on 31 January 2022 to address its limitations and harmonize the rules governing clinical trials across all EU member states, making them binding and uniform.
What was the reason for the transition from EU CTD to EU CTR?
The transition from the EU Clinical Trials Directive (EU CTD; 2001/20/EC) to the EU Clinical Trials Regulation (EU CTR; Regulation (EU) No 536/2014) occurred to address several critical shortcomings in the previous framework and to modernize the conduct of clinical trials across the European Union.

Key Reasons for the Transition
1: Fragmentation and Inconsistency
The EU CTD was a directive, meaning each member state interpreted and implemented it differently, leading to fragmented processes, inconsistent requirements, and duplicated work for multinational trials.
2: Excessive Administrative Burden for Sponsors
Sponsors had to file separate applications for each country involved in a trial, resulting in a heavy administrative load, increased costs, and delays in trial authorization.
3: Lack of Harmonization
Ethics and regulatory reviews varied across jurisdictions, slowing down the approval and conduct of clinical trials especially multi-country studies.
4: Reduced Competitiveness
The complex regulatory environment under the CTD caused a decline in the number of clinical trials conducted in the EU, as sponsors shifted studies to more efficient regions.
5: Insufficient Transparency
The CTD did not mandate public access to all clinical trial data, limiting transparency and public trust in the EU clinical trials system.
What are the key changes made from CTD and CTR?
Key changes introduced by the EU Clinical Trials Regulation (CTR) include:
1: All new trial applications must be submitted via CTIS, the centralized online portal for managing clinical trials across all EU/EEA member states.
A transition period is in effect until January 31, 2025, after which all ongoing trials must fully comply with the CTR framework via CTIS.
2: Sponsors can submit a single application that undergoes a coordinated assessment by all concerned member states, simplifying multinational trial approvals.
3: Defined and coordinated timelines for evaluation and approval to speed up clinical trial authorization across countries.
4: Public access to clinical trial data, including applications, protocols, and results, is required, increasing transparency and public trust.
5: Safety reporting requirements have been standardized and reinforced, ensuring stronger protection for trial participants.
6: By reducing regulatory complexity, the CTR supports the smooth conduct of multinational trials, making it easier for sponsors to launch and manage studies across multiple countries.
7: Regulatory authorities and ethics committees now collaborate more closely within the CTR framework, promoting greater consistency and coordination in trial supervision.
Who Does and What Does the EU CTR Apply To?
The EU Clinical Trial Regulation (CTR) applies broadly to organizations and individuals involved in the conduct of clinical trials within the European Economic Area (EEA), including all EU member states. Its scope and impact are defined as follows:
1: Clinical Trial Sponsors
Any organization or individual (commonly pharmaceutical companies, biotech firms, academic institutions, or independent investigators) that initiates, manages, and finances a clinical trial of medicinal products in the EU/EEA.
2: Investigators and Research Sites
Principal investigators and institutions conducting the trial at clinical sites based in the EU/EEA.
3: Contract Research Organizations (CROs)
Entities acting on behalf of sponsors to carry out trial-related duties and functions.
Also read: The Role of CROs in Managing Multilingual Clinical Trial Documentation
What does the EU CTR apply to
1: All Interventional Clinical Trials
Applies to any clinical trial, whether national or multinational, where the effects, safety, or pharmacology of a medicinal product are being investigated and the participant is subjected to protocol-driven intervention or modifications from standard clinical care.
2: Both National and Multi-country Trials
Covers trials conducted in a single EU/EEA country as well as those run in multiple countries within the EU/EEA.
3: Low-Intervention Trials
Also regulates low-intervention clinical trials with provisions for adapted rules
The EU CTR does not apply to:
1: Non-interventional (Observational) Studies
Studies where participants receive treatments as per normal clinical practice, with no protocol-specified intervention, randomization, or added tests/procedures beyond routine care.
2: Trials Outside the EU/EEA
The regulation exclusively governs trials conducted within the EU/EEA; trials conducted elsewhere are not covered.
Are You Looking for professional Clinical Trial Translations?
Language and Translation Requirements Under the EU CTR

The EU Clinical Trial Regulation (CTR) 536/2014 establishes harmonized, mandatory language and translation rules for clinical trials involving medicinal products in the EU and EEA.
Its focus is on enhancing participant protection, ensuring regulatory efficiency, and fostering transparency across all member states.
1: Application Language
Each Member State Concerned (MSC) sets its own language requirements for application submissions. While many accept application documents in English, some require translations into their official national language(s).
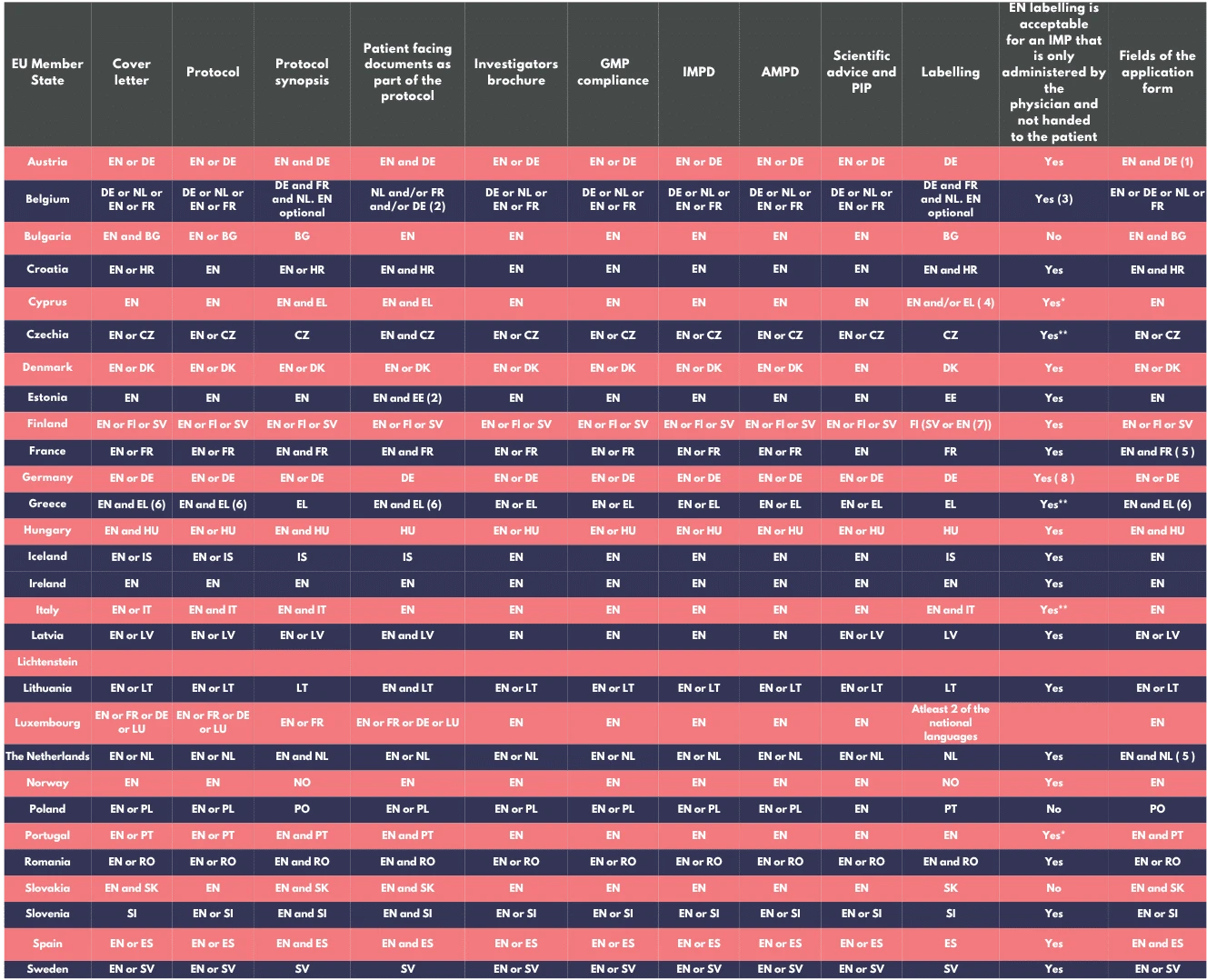
2: Part I Documents (Scientific & Medicinal Product Information)
These documents are generally submitted in English, but if requested by an MSC, translations into the local language must be provided.
3: Part II Documents (National Specific Information)
Must be submitted in accordance with the official language(s) of each MSC, as they include nationally specific elements such as recruitment practices, informed consent content, and local regulations.
4: Documents for Trial Participants
All participant-facing documents (e.g., informed consent forms, patient information sheets, questionnaires) must be translated into the official language(s) spoken by trial participants to ensure comprehension and informed consent.
Also read: ICF Translation: Importance, Requirements & Best Practices
5: Translation Quality
Translations must be accurate and done by professional translators with expertise in medical/scientific terminology. Sponsors should ensure quality control processes to maintain consistency and clarity.
6: Layperson Summaries
Clinical trial results summaries intended for the general public are required to be translated into the official language(s) of the member states where the trial took place, promoting transparency and accessibility.
7: Record Keeping
Sponsors are expected to keep detailed records of translations, including version history and translator qualifications, to comply with regulatory inspections and audits.
8: Variability and Clarifications
Since language requirements, especially for national-specific documents, vary by member state, it is advisable for sponsors to consult individual MSC guidelines early in the trial preparation to avoid delays.
Best practices for clinical trial translation

1: Partner with a Certified Translation Agency
Choose an agency that has ISO 17100 and ISO 9001 certification, this shows they have the expertise in healthcare translations.
These agencies work with trained medical linguists who understand both complex medical terminology and regulatory requirements. Their expertise helps prevent costly errors that could compromise patient safety or delay trial approvals.
2: Collaborate with Medical Experts
Working healthcare professionals in the translation process ensure medical accuracy and contextual relevance.
Translators bring linguistic and subject-matter expertise, while medical experts validate terminology and clinical concepts. This collaboration is essential for maintaining scientific integrity and regulatory compliance.
3: Use Patient-Centric Language
Clinical trial materials must be easy for participants to understand. Simplifying medical jargon into clear, patient-friendly language improves comprehension and supports informed consent.
4: Quality Assurance Process
Implementing a robust quality assurance process is a must in clinical trial translation. A reliable QA process includes steps, such as reviews, edits, and back translations are necessary for ensuring consistency.
5: Maintain Confidentiality
Protecting the privacy of patients as well as ensuring that any sensitive trial data is preserved is a top priority and among the most important translation requirements for clinical trials.
To achieve robust security measures, translators must adhere to strict confidentiality protocols to safeguard personal and clinical information. These guidelines should be in compliance with global data protection standards.
6: Ensure Consistency with CAT Tools and Glossaries
Consistency in terminology is essential across clinical trial documents to avoid confusion and ensure clarity. Tools like CAT software, termbases, and translation memories help maintain uniformity across multiple languages and file formats.
7: Maintain Records
Keeping detailed records of each step in the translation process—including edits, reviewer comments, and approvals—is essential for transparency and compliance. These records ensure traceability, support regulatory audits, and help resolve issues efficiently, keeping all stakeholders aligned.
Conclusion
The EU Clinical Trials Regulation (CTR) establishes a unified legal framework for conducting clinical trials across EU/EEA member states. As part of this harmonized approach, the regulation emphasizes the importance of clear communication through accurate and accessible translations of trial documentation.
Sponsors are required to provide key documents in the official languages of the countries where the trial is conducted.
Meeting these language requirements is not only a regulatory obligation but also a critical factor in safeguarding participant comprehension, upholding ethical standards, and ensuring successful trial implementation across borders.
With deep expertise in life sciences and a network of native-language experts, Milestone Localization is your trusted partner for delivering accurate and compliant clinical trial translations that meet EU regulatory standards and support the success of your global research initiatives.
Also read: Translating Clinical Trials: Global Implications and Responsibilities
Get Your Clinical Trials Translated
FAQS
What is the primary purpose of translation requirements under EU CTR 536/2014?
To ensure clear, accurate communication for trial participants and regulatory authorities, enhancing participant protection, regulatory efficiency, and transparency.
Which documents require translation, and in which languages must they be submitted?
-
Part I documents (scientific and medicinal product info) are usually in English but may need local language translation upon request.
-
Part II documents (national-specific info) must be in official languages of each member state concerned.
-
Participant-facing documents (e.g., informed consent forms) must be in the language(s) spoken by the participants.
Who should perform translations for clinical trials under EU CTR 536/2014?
Translations must be done by professional translators with expertise in medical and scientific terminology, ideally working with healthcare experts for accuracy.
Are there quality requirements for translations under the EU CTR?
Yes, translations must be accurate, and sponsors are encouraged to implement quality assurance processes including reviews, edits, and back translations.
How should sponsors handle translation records and confidentiality?
Sponsors must keep detailed records of translation versions and translator qualifications and ensure strict confidentiality to protect patient data.
Do language requirements vary between EU member states?
Yes, language requirements, especially for national-specific documents, vary by member state, so it is advisable to consult individual member state guidelines during trial preparation.