")

The UK’s clinical trial framework is governed by the Medicines and Healthcare products Regulatory Agency (MHRA), which ensures that studies are conducted safely, ethically, and transparently.
A key aspect of this framework is its focus on clear and accurate language so that both regulators and trial participants fully understand all trial-related information.
To achieve this, the MHRA sets expectations for how information should be written and when needed, translated to meet regulatory standards.
This blog highlights the MHRA’s language requirements for clinical trials in the UK, the implications of the new 2025 MHRA Clinical Trials Reform, and best practices for clinical trial translations.
What is MHRA?
The MHRA (Medicines and Healthcare products Regulatory Agency) is an executive agency of the UK’s Department of Health and Social Care responsible for regulating medicines, medical devices, and blood components for transfusion within the United Kingdom.
It ensures that medicines and medical devices are safe, effective, and meet quality and regulatory standards before they can be prescribed or sold in the UK.
What does the MHRA say about Clinical Trials?
The MHRA regulates how clinical trials are designed, approved, and conducted in the UK to ensure participant safety and credible results.
Before a study begins, the MHRA must authorize the trial, confirming that the medicine or device is suitable for human research and that the protocol protects participants.
Trials must also receive approval from a Research Ethics Committee and follow Good Clinical Practice (GCP).
During the study, the MHRA reviews safety reports and can pause or stop a trial if the risks outweigh potential benefits. Overall, the MHRA’s oversight ensures trials are ethical, well-controlled, and reliable.
Overview of the 2025 MHRA Clinical Trials Reform
The 2025 MHRA Clinical Trials Reform is the UK’s most significant update to clinical trial regulations in over 20 years. The Medicines for Human Use (Clinical Trials) (Amendment) Regulations 2025, signed into law in April 2025, will take full effect on April 28, 2026, after a 12-month transition.
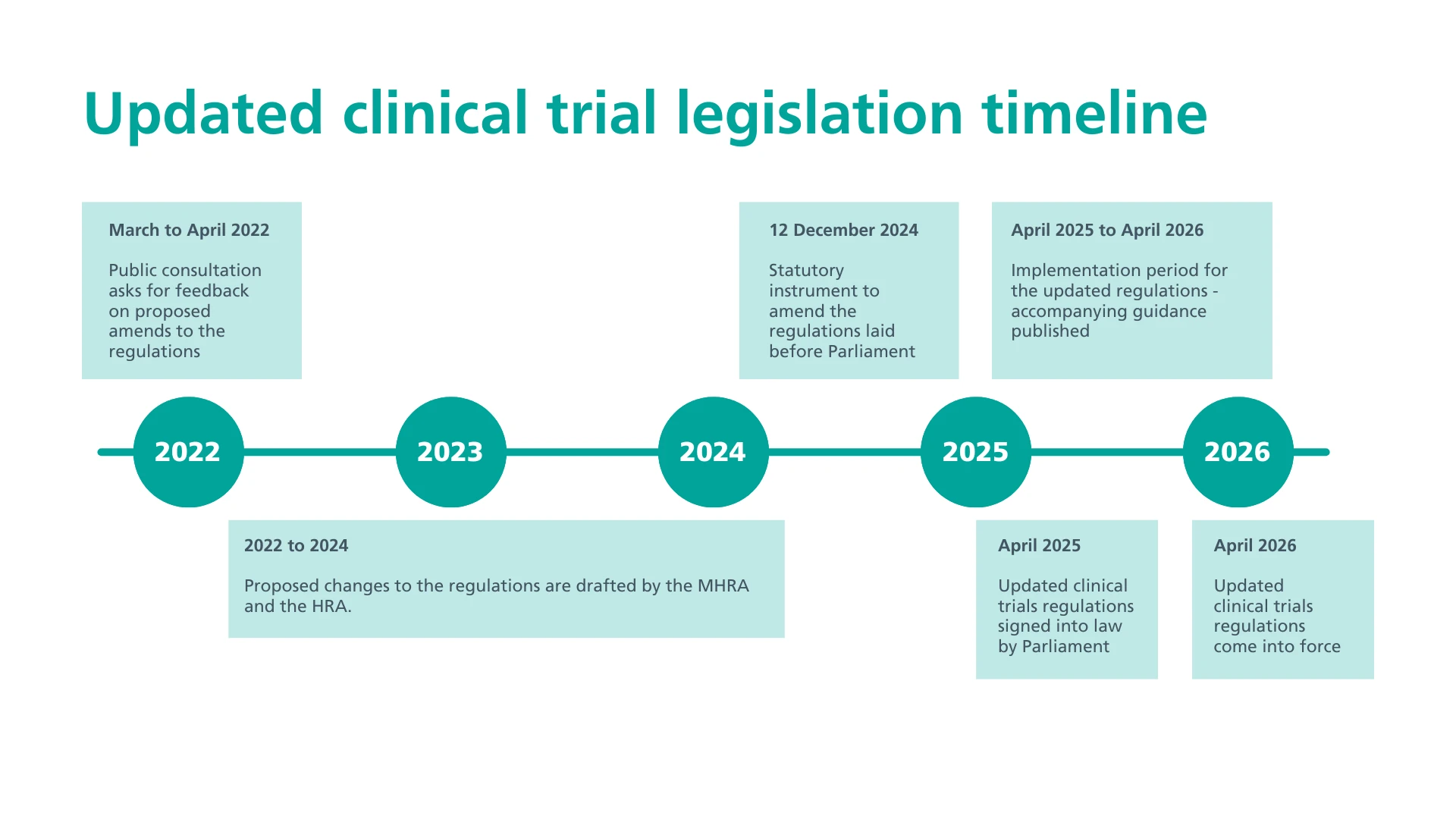
The reform aims to make the UK a faster, more flexible, and attractive place for high-quality research, speeding up new treatment development while ensuring patient safety.
Here are the key changes in the new MHRA framework:
A: Streamlined and Risk-Proportionate Approvals
1: Combined Review
The combined regulatory and ethics review is now formally embedded in law, creating a single, coordinated decision process.
2: Review Timelines
After a 7-day validation period, the combined decision will be issued within a maximum of 30 calendar days for most applications. The time to respond to a Request for Further Information (RFI) is also extended to 60 days, aligning UK trial timelines more closely with international standards.
3: Proportionality
The reform introduces a risk-based system, including “Notifiable Trials” for certain lower-risk studies, such as those using approved medicines.
4: Trial Lifetime
A sunset clause is now in place, meaning trial approval will lapse after 2 years if no participants have been recruited.
B: Enhanced Transparency and Patient-Centricity
1: Mandatory Public Registration
All clinical trials must now be registered in a WHO-compliant public registry.
2: Mandatory Results Publication
A summary of trial results must be published within 12 months of trial completion. Limited deferrals are allowed to protect commercially confidential information.
3: Participant Summary
Sponsors are required to offer participants a clear, easy-to-understand summary of trial results.
4: Simplified Consent
Certain lower-risk trials, such as those using established medicines, can use simplified consent processes.
C: Good Clinical Practice (GCP) & Pharmacovigilance
1: Updated GCP
The 2025 regulations require compliance with ICH-GCP E6(R3) to ensure UK trial data is globally accepted. This provides a modern framework for conducting clinical trials.
2: Terminology Alignment
Key terms are updated, such as ‘amendment’ → ‘modification’ and ‘subject’ → ‘participant’. This ensures consistency with international standards.
3: Safety Reporting Streamlining
Sponsors are no longer required to include detailed listings of Serious Adverse Events (SAEs) or Serious Adverse Reactions (SARs) in their annual Development Safety Update Reports (DSURs) submitted to the MHRA and RECs.
Also read: The Role of CROs in Managing Multilingual Clinical Trial Documentation
Language and Translation Requirements Under the New MHRA Framework

Under the 2025 MHRA Reform, Sponsors are required to communicate trial information and results in a way that is clear, plain, and easy for participants to understand.
Below is an overview of the key language and translation requirements under the new framework.
1: Participant Information and Consent Forms (PIS/CF)
All documents provided to participants must be clear, written in plain language, and easy to understand. If participants do not speak English, translations are legally required to ensure valid and ethical consent.
2: Simplified Consent Arrangements
For certain low-risk trials, simplified consent is allowed, but information must remain understandable and jargon-free.
3: Operational Implication
The 30-day approval clock for combined MHRA/Ethics review means all translated PIS/CFs must be ready before submission.
4: Mandatory Lay Summary of Results
Sponsors must provide participants with a summary in a format they can understand. If participants speak other languages, translations are required to meet legal obligations.
For the full scope of regulated documentation, our medical device translation services cover everything from IFUs to clinical trial materials.
5: Public Registry Publication
Trial results must be published within 12 months in a public registry. While English is standard, multilingual summaries are encouraged to enhance global accessibility and transparency.
All translated materials must be version-controlled and documented in the Trial Master File (TMF) to demonstrate compliance.
Also read: Translating Clinical Trials: Global Implications and Responsibilities
Looking for clinical Translation services?
Impact of the 150-Day Timeline on Translation Workflows
The 150-day UK clinical trial set-up target requires Sponsors and CROs to integrate translation early in the study start-up process. Translation can no longer wait until after approvals; it must run in parallel with protocol finalization and the Combined Review submission.
All participant-facing materials (PIS/ICF, recruitment content, etc.) must be submitted in final translated form upfront, as ethics committees now assess both content and language accessibility from the outset.
As a result, Sponsors must:
1: Plan translation early, alongside regulatory and operational planning.
2: Finalize English source documents sooner to allow time for accurate multilingual preparation.
3: Adopt efficient translation workflows and technology (TMS, MT+post-editing, terminology management).
4: Identify required languages upfront to avoid timeline disruptions.
5: Coordinate early with CROs and translation partners to maintain consistency and speed.
What Should Ethics Committees Evaluate Now
1: Informed Consent & Accessibility
Ensuring PIS/ICFs are written in plain language and accessible, including plans for translations where needed. They also assess ethical justification for simplified consent in low-risk trials.
2: Inclusion & Diversity
Reviewing the Sponsor’s Inclusion and Diversity Plan to ensure recruitment reflects the population relevant to the condition being studied.
3: Transparency & Post-Trial Results
Confirming plans for trial registration, timely results publication, and providing Lay Summaries to participants in a language they can understand.
4: Risk-Proportionate Review Efficiency
Working with MHRA to meet the 30-day decision timeline, issuing RFIs only where necessary, and avoiding duplication of MHRA scientific review.
Best Practices For Clinical Trial Translation

1: Partner with a Certified Translation Agency
Choose an agency that has ISO 17100 and ISO 9001 certification, which shows that they have well-documented processes.
These agencies work with trained medical linguists who understand both complex medical terminology and regulatory requirements. Their expertise helps prevent costly errors that could compromise patient safety or delay trial approvals.
2: Collaborate with Medical Experts
Working healthcare professionals in the translation process ensure medical accuracy and contextual relevance.
Translators bring linguistic and subject-matter expertise, while medical experts validate terminology and clinical concepts. This collaboration is essential for maintaining scientific integrity and regulatory compliance.
3: Use Patient-Centric Language
Clinical trial materials must be easy for participants to understand. Simplifying medical jargon into clear, patient-friendly language improves comprehension and supports informed consent.
4: Quality Assurance Process
Implementing a robust quality assurance process is a must in clinical trial translation. A reliable QA process includes steps, such as reviews, edits, and back translations are necessary for ensuring consistency.
5: Maintain Confidentiality
Protecting the privacy of patients as well as ensuring that any sensitive trial data is preserved is a top priority and among the most important translation requirements for clinical trials.
To achieve robust security measures, translators must adhere to strict confidentiality protocols to safeguard personal and clinical information. These guidelines should be in compliance with global data protection standards.
6: Ensure Consistency with CAT Tools and Glossaries
Consistency in terminology is essential across clinical trial documents to avoid confusion and ensure clarity. Tools like CAT software, termbases, and translation memories help maintain uniformity across multiple languages and file formats.
7: Maintain Records
Keeping detailed records of each step in the translation process including edits, reviewer comments, and approvals is essential for transparency and compliance. These records ensure traceability, support regulatory audits, and help resolve issues efficiently, keeping all stakeholders aligned.
Conclusion
The UK’s MHRA framework places strong emphasis on clarity, transparency, and patient safety throughout the clinical trial process.
To meet these expectations, sponsors must ensure that all participant-facing and regulatory documents are accurate, accessible, and presented in language that is clearly understood by local stakeholders.
Fulfilling these language and documentation requirements is not only a regulatory necessity but also essential to maintaining ethical standards, informed participation, and successful trial delivery within the UK.
Also read: EU CTR 536/2014: Translation Requirements
Are You Looking for Certified clinical Translation services?
FAQS
Why are language requirements important for clinical trial documentation in the UK?
The MHRA requires that all study documents are clear, accurate, and understandable to ensure participant safety and informed consent. Documents must be written in a language that participants can comfortably understand to avoid misunderstandings and ensure ethical compliance.
Which clinical trial documents must be translated under MHRA regulations?
Key materials such as Patient Information Sheets (PIS), Informed Consent Forms (ICF), recruitment materials, questionnaires, and participant-facing instructions must be provided in a language that the participant can understand. Technical and regulatory documents may remain in English unless otherwise required by stakeholders.
Does the MHRA specify which languages must be used?
No. The MHRA does not mandate specific languages. However, sponsors must ensure that materials match the linguistic needs of the participant population—e.g., translating documents for multilingual communities or non-native English-speaking participants.
Who is responsible for ensuring the accuracy of translations?
The trial sponsor holds responsibility. They are expected to use qualified medical translators and, when needed, back-translation and review by clinical experts to ensure medical accuracy and consistency.
Do translated documents need to be approved by the Ethics Committee?
Yes. All participant-facing translated materials must be submitted to the Research Ethics Committee (REC) for review and approval before they are used in the trial.